ゲノムサイズが未知で、Whole-genomeを使っての集団解析を行うのは厳しい、それでも解析を行いたいというときに活躍する方法としてRAD-seq(Restriction-site Associated DNA Sequencing)という方法を紹介します。
RAD-Seqは次世代シーケンシング技術を用いた、制限酵素認識サイトの近隣領域(ゲノム全体の0.1~1%程度)を解析する手法です。
この方法では下記の図のようにゲノムを制限酵素で切断し、切断した部位から50-100bp程度のシーケンスを読み、SNPsを探索します。
シーケンス範囲が狭いため、通常のWhole genomeをショットガンシーケンスで読むよりも低コストで効率よく解析が行えます。
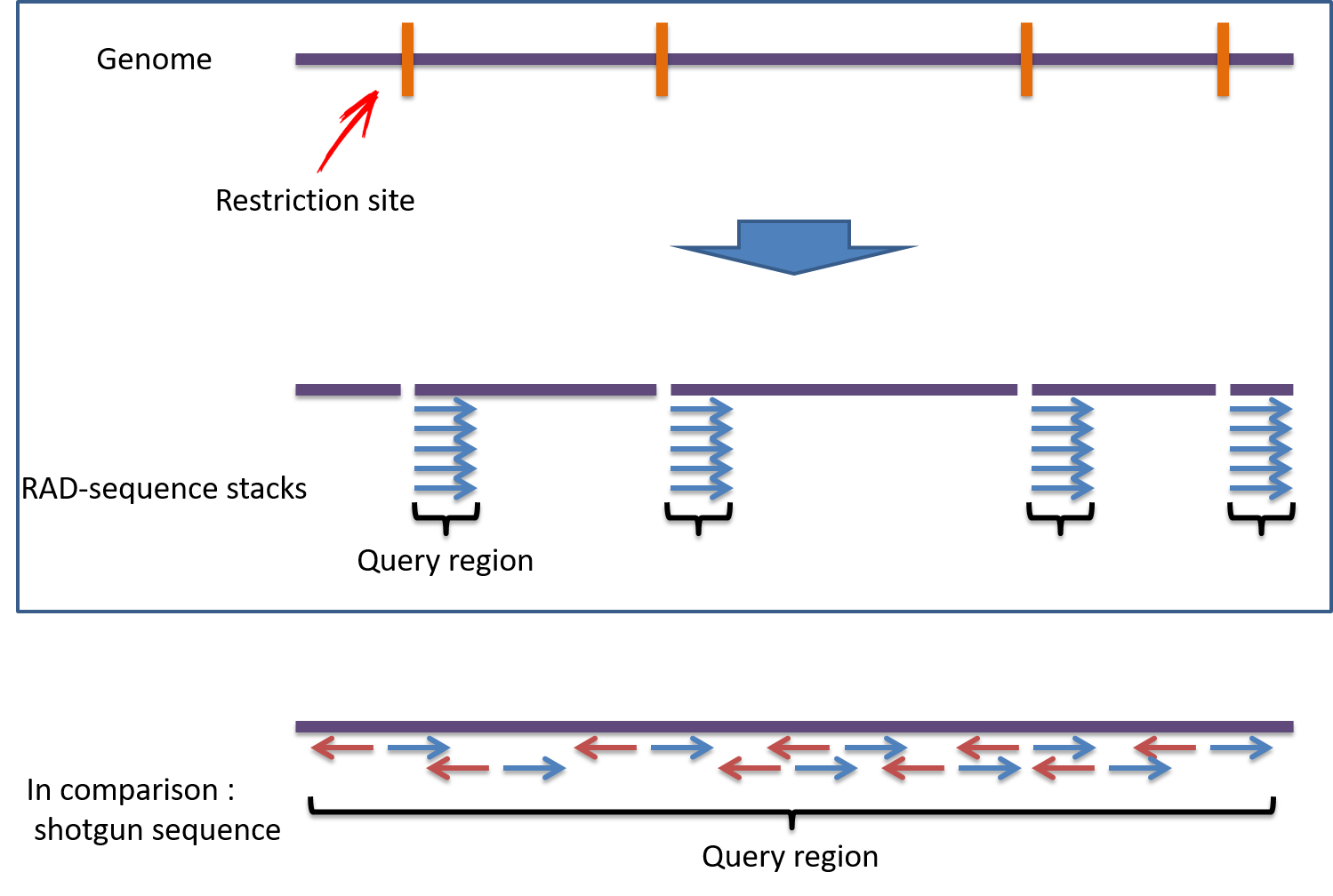
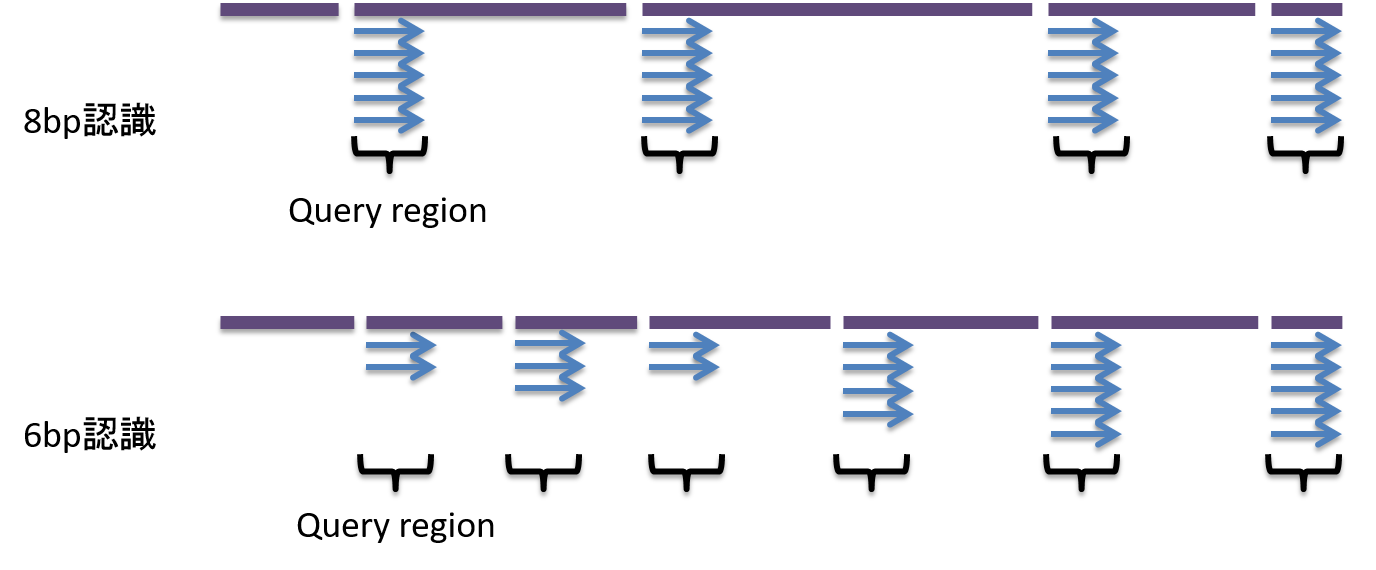
また制限酵素の種類を変えることで、高密度解析、低密度解析の調整が可能となります。
低密度解析: 低コスト、探索範囲は狭い (e.g. 8bp認識の制限酵素カット)
高密度解析: 高コスト、探索範囲は広い (e.g. 6bp認識の制限酵素カット)
ただいろいろやってみた感想ですが、ゲノムサイズが大きすぎると制限酵素サイトもそれに応じて大きくなるので、カバレッジが低くなる傾向があるようです。
変異が生じていると断ずるには最低5カバレッジは欲しいところです。カバレッジが低くても、候補を増やしたい時には、ツールの方でカバレッジを低くして、よりセンシティブに検出できるようにする方法があります。・・・が、あまりお勧めしません。一度解析して、いい結果が出そうであれば、素直にもう一度読んでデータ量を増やしたほうがいいと思います。
使える解析としては、
・種内での分子系統解析
・品種・系統・集団間での遺伝的変異の同定
・集団構造解析
・連鎖解析
・特定の遺伝子のマッピング
・QTL解析
(Peterson et al., 2012)
など。
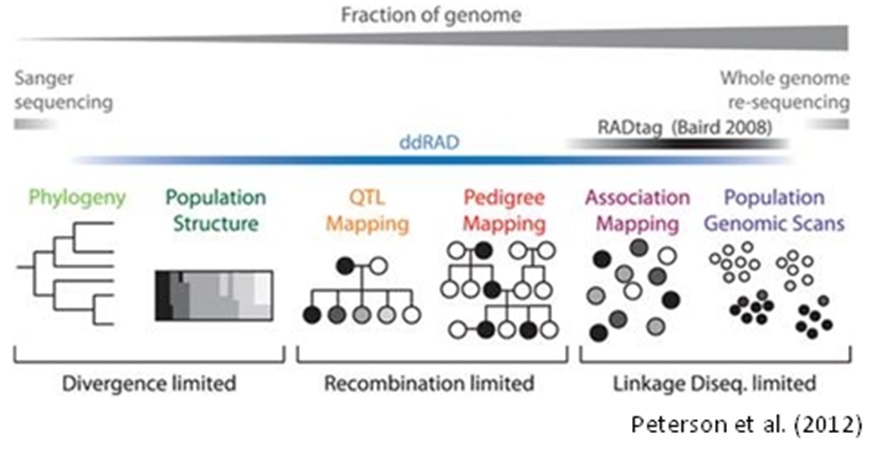
上の使える解析をみて、あれ?そういえば似たようなことが分かる解析があったぞ。って思った方正解です。
ISSR(Inter Simple Sequence Repeat)で行う解析も同じようなことが出来ましたね。
これらを利用したMIG-Seq(multiplexed ISSR genotyping by sequencing)という解析手法もあります。
考え方としてはRAD-seqと同じで、制限酵素の部分がISSR認識のプライマーに変わった手法となります。
http://www.nature.com/articles/srep16963
解析ソフトとしては有名なものに、 ・Stacks ・pyRAD があります。 pyRADのほうがInDelを考慮してくれる反面、そのせいで解析時間が結構かかるという特徴があります。また使ってみた個人的感想として、1サンプル当たりの配列数が少ないと、Stacksと比較して、データが出にくい傾向があるように思います。
受託解析や共同研究では、上のStacksを使うことの方が多く論文もStacksのほうが多いようです。
最後にコツですが、
まず制限酵素選びが重要です。先ほど6塩基認識、8塩基認識のところでも説明しましたが、探索範囲が大幅に変わってきます。生物種によっても異なるため、一度少量だけでも読んでみて、検討してみたほうがいいと思います。
こちらにわかりやすくまとまっておりますので、確認してみてください。
https://www.ddbj.nig.ac.jp/wp-content/downloads/ddbjing/33ddbjing_shirasawa.pdf#page=8
次回は実際にStacksを使った解析手法についてご紹介したいと思います。
