シーケンサーはホモポリマーを読むのが苦手という弱点があります。
次世代シーケンサーのMiSeq はこの点に強いと紹介されており(こちら)、我々も実際にMiseqのデータを扱うことが多いので今回ホモポリマーに着目してみました。
サンプルは、ある生物(クローン)の腸内 18S メタゲノムです。
解析の際、ホストのゲノムが大量に得られました。ホストはクローンのため、理論的には得られたホストゲノムは配列が一致します。データ処理はトリミング、アセンブル(ペアリードマージ)、キメラ除去を行った後、Unique(100%一致する配列をまとめる処理)をして それぞれのまとまりをOTU とし、カウントを行いました。
結果、 OTU が大量に生成されました。配列を見てみると、数塩基の違いでまとまらなかったことが原因のようです。
そこで、具体的にどういったところにシーケンスエラーが起こりやすいか、確認してみました。リードは読み始め、読み終わりはクオリティが下がる傾向にあるため、中間のマルチプルアライメントを取り出してみます。

円でしるしを付けた箇所がシーケンスエラーの可能性が高い配列です。傾向としては、連続した塩基やその周囲で起こっているように見られます。
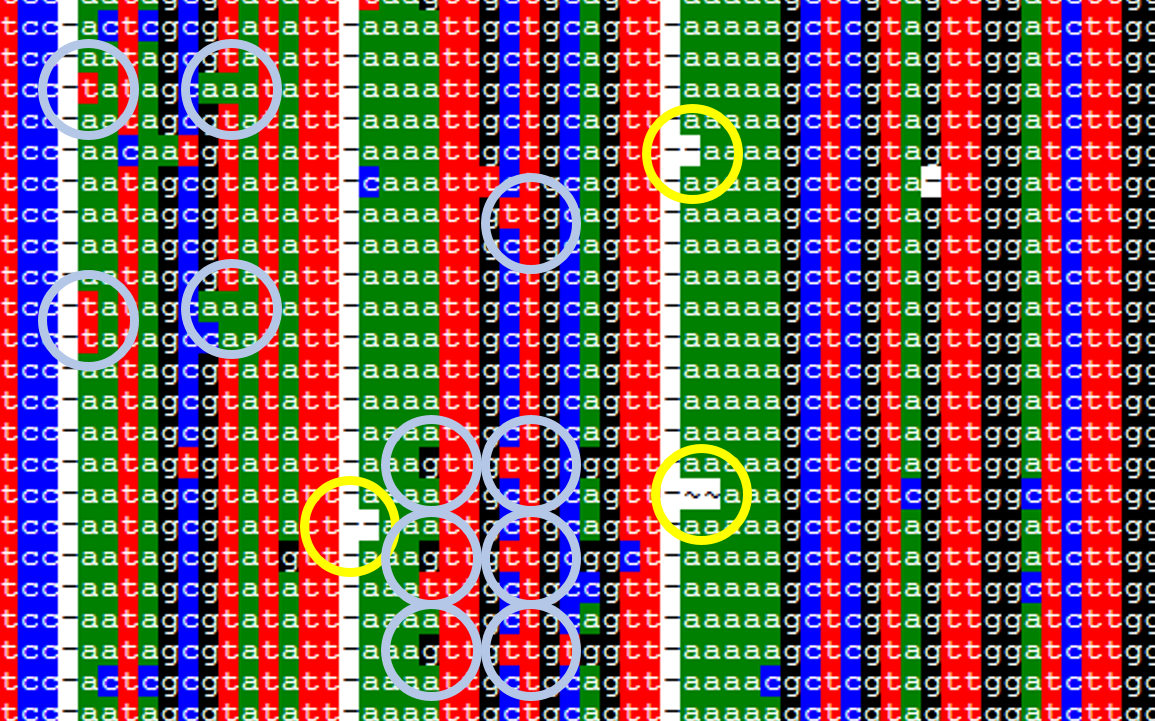
同じく繰り返し配列やその周辺に見られる様子です。水色で示している箇所は同じ部位に複数見られたものを表しています。
配列を得るまでのどの段階でこのようになるかはわかりませんが、情報解析時にはこれらの結果をふまえて計算することになります。ではメタゲノム解析でクラスタリングする際、いくつまでのミスマッチを許容すべきなのでしょうか。
Unique にした時点のホスト OTU 数が 983 あったのですが、この個体はクローンなので “1” OTU にまとまることを目指します。
Simirality 基準で OTU を段階的にまとめてみます。縦軸に OTU 数、横軸に Simirality をまとめる際の値をとると、以下のグラフになります。
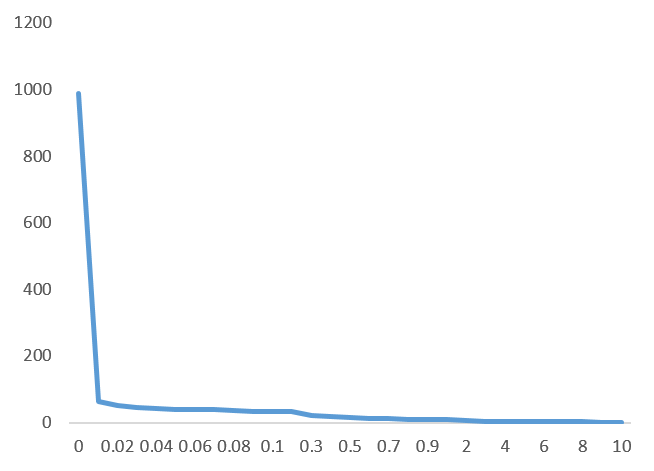
フラグメントサイズ 420bp のサンプルですが、グラフから Simirality 0.91 の時点で一つの OTU になりそうです。38bp 、または、10% までミスマッチを許容する設定になります。
かなり緩い設定となってしまいますが、ざっくりとした優占種を調べたいときは、影響は少ないかと思います。一方、レアな個体や、ある種に近い未知の個体を調べる際には、問題が出てきてしまう可能性があります。用途によってこの値を調整することが重要と考えられます。